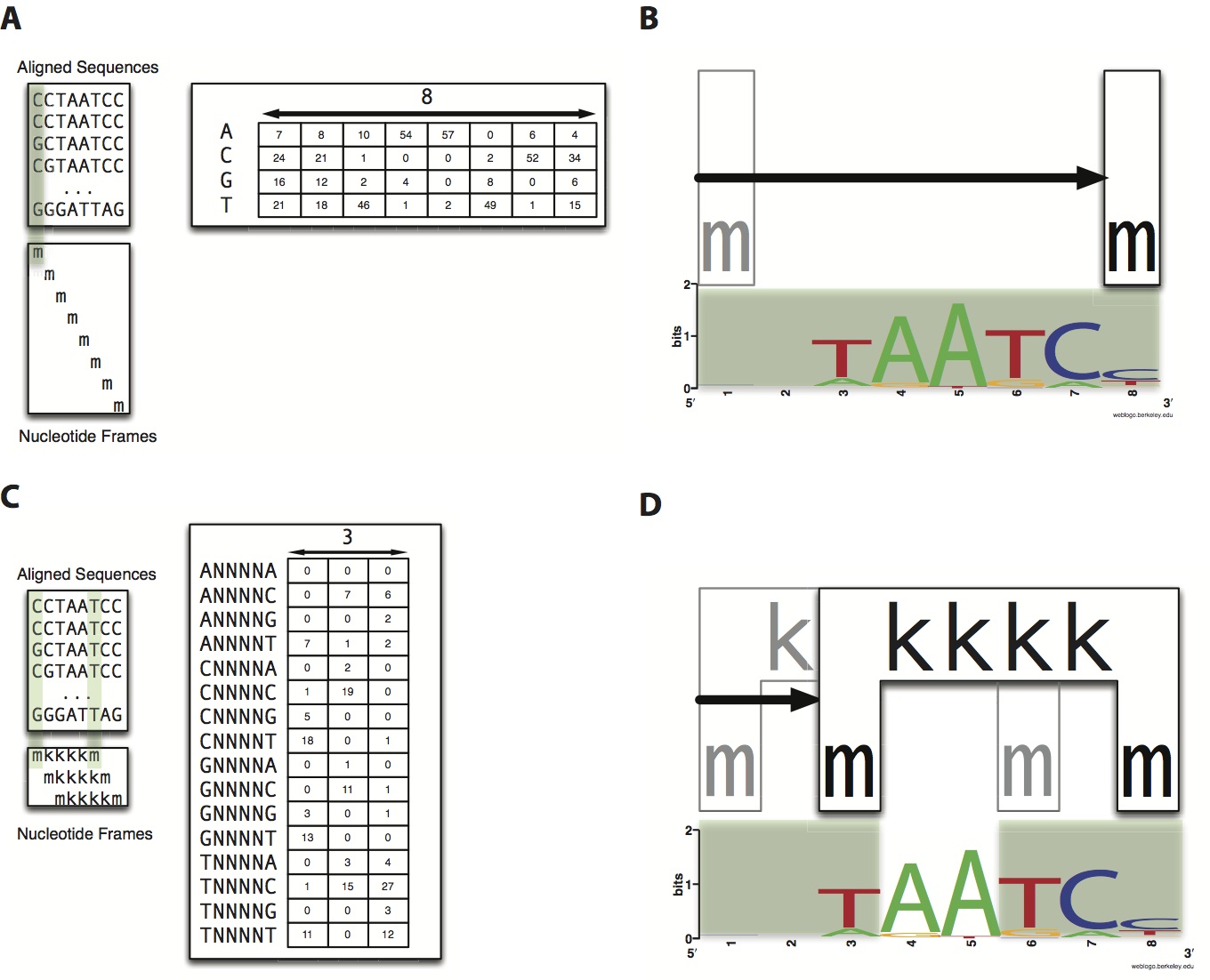
Predicting the location of TF binding sites within a genome is a difficult task, but is an integral part in furthering our understanding of gene regulation. Traditional methods involve PWMs (position weight matrices), which rely on the underlying assumption that each nucleotide within a TF binding site is independent of the other nucleotides. More recently, models have been implemented which relax this assumption to include dependence on neighboring nucleotides (i.e. a string of contiguous nucleotides).
We have developed a novel algorithm, MARZ, which not only includes dependence on adjacent nucleotides, but allows for gaps in dependence. This unbiased computational algorithm systematically analyzes all possible gapped matrices across a fixed number of nucleotides. Our preliminary results indicate that in many cases gapped matrix models can outperform traditional models, but that the relative strength of the binding sites considered in the analysis also plays a role in their predictive ability.