We study how evolutionary forces shape the genome and how these changes influence phenotype. To address this topic we heavily utilize next-generation sequencing and combine comparative, functional, and population genomics.
Fungal Domestication
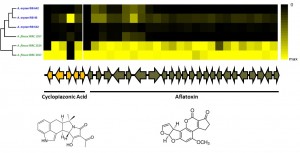 For thousands of years, humans have utilized various fungal species for use as fermenting agents, flavor enhancers, and food sources (think wine, cheese, shiitake mushrooms etc.). During this process, humans propagated isolates with specific desired characteristics, and in doing so, altered these fungal genomes. Domestication genomics offers an excellent model to understand the influence of selection on the genome. We employ population genomics and functional genomics to identify regions of the genome shaped by the domestication process.
For thousands of years, humans have utilized various fungal species for use as fermenting agents, flavor enhancers, and food sources (think wine, cheese, shiitake mushrooms etc.). During this process, humans propagated isolates with specific desired characteristics, and in doing so, altered these fungal genomes. Domestication genomics offers an excellent model to understand the influence of selection on the genome. We employ population genomics and functional genomics to identify regions of the genome shaped by the domestication process.
Fungal Pathogenicity
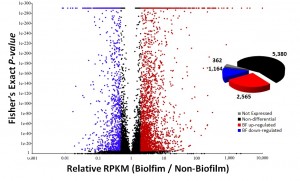 Opportunistic fungal pathogens are an emerging threat to human health. Pinpointing the genetic toolkit and the functional mechanisms which allow fungi to invade and colonize host tissue is essential for the their treatment and control. We interrogate genome sequencing data and gene expression data to identify genomic regions underlying pathogenicity.
Opportunistic fungal pathogens are an emerging threat to human health. Pinpointing the genetic toolkit and the functional mechanisms which allow fungi to invade and colonize host tissue is essential for the their treatment and control. We interrogate genome sequencing data and gene expression data to identify genomic regions underlying pathogenicity.
Copy Number Variation
Copy Number Var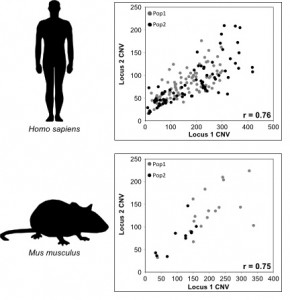 iation (CNV) is widespread across eukaryotic genomes and is an important source of genetic diversity and phenotypic variation. In the past, due to technical limitations, CNV was difficult to identify. However, advances in DNA sequencing technologies and computational methods have greatly improved CNV detection. We use next-generation sequencing data to characterize (a) population-level patterns and (b) functional significance of CNV.
iation (CNV) is widespread across eukaryotic genomes and is an important source of genetic diversity and phenotypic variation. In the past, due to technical limitations, CNV was difficult to identify. However, advances in DNA sequencing technologies and computational methods have greatly improved CNV detection. We use next-generation sequencing data to characterize (a) population-level patterns and (b) functional significance of CNV.