
Our current research focuses on computer simulations of protein allosteric effect, intrinsically disordered proteins, and amyloid aggregate-membrane interactions.
Ongoing projects
1. Protein misfolding and type II diabetes
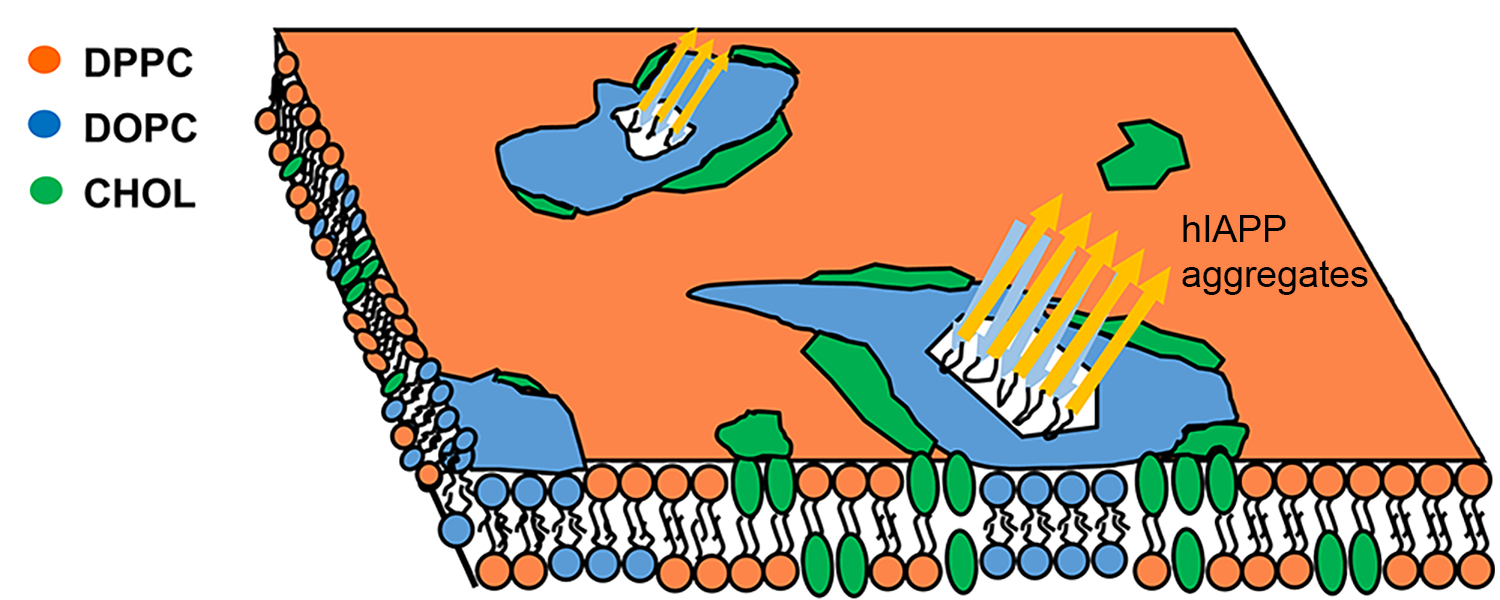 hIAPP insertion affects the raft-containing membranes
hIAPP insertion affects the raft-containing membranes
Human islet amyloid polypeptides (hIAPP) aggregate into amyloid deposits in the pancreatic islets of Langerhans, contributing to the loss of β-cells in patients with type 2 diabetes. Despite extensive studies of membrane disruption associated with hIAPP aggregates, the molecular details regarding the complex interplay between hIAPP aggregates and raft-containing membranes are still very limited. Using all-atom molecular dynamics simulations, we investigate the impact of hIAPP aggregate insertion on lipid segregation. Our investigations on the interaction between hIAPP aggregates and raft-containing membranes could lead to a better understanding of the mechanisms of amyloid cytotoxicity. We also collaborate with the Jakobsche group in our department to study the properties of the third-generation amyloid-binding bifunctional molecules that can bind to amyloid fibrils and modify the properties of those fibrils by delivering various functional molecular cargos to them.
2. RING-type E3 ubiquitin ligase
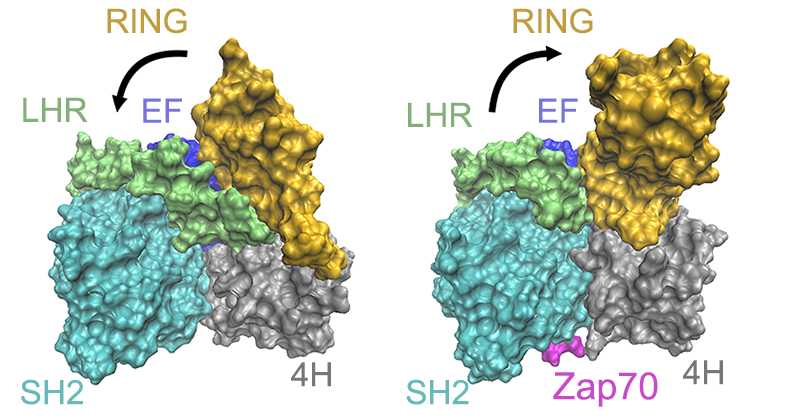 The binding of Zap70 shifts the RING domain to the partially-open state
The binding of Zap70 shifts the RING domain to the partially-open state
Ubiquitin serves as a tag to selectively label proteins for degradation. E1, E2, and E3 ligases are enzymes acting sequentially in the protein degradation cascade to transfer ubiquitin to the target protein to be degraded. A deep understanding of protein degradation mechanisms will help to design drugs to selectively degrade disease-causing proteins, ranging from those associated with Alzheimer’s disease to cancers. c-Cbl is one of the RING-type E3 ligases. X-ray structures show that in the absence of E2 ligase, c-Cbl adopts a closed conformation before it binds with its substrate. This closed form is considered to be unactivated in the degradation cascade. While after the substrate binding, the RING domain is partially open, which is the first step of activation of c-Cbl. As a first step to elucidate the details of the activation mechanism, computer simulations have been used to investigate the dynamic features of the closed state of c-Cbl and its partially opened state as well as the allosteric effect. Our simulations will provide new insights into the critical role of protein conformational changes in the initial steps of c-Cbl activation.
3. Algorithm Development
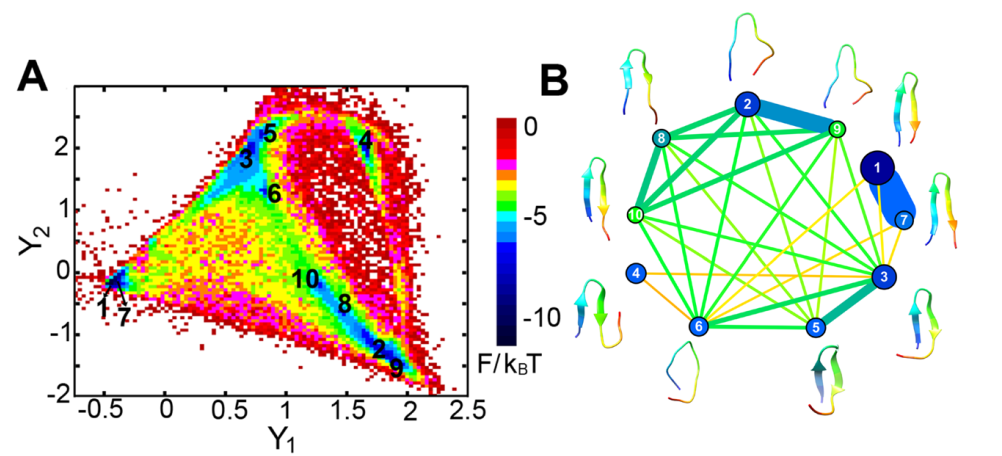 Left panel: Diffusion map embedding of the free energy landscape of β-hairpin; Right panel: Flow network of β-hairpin conformational transitions
Left panel: Diffusion map embedding of the free energy landscape of β-hairpin; Right panel: Flow network of β-hairpin conformational transitions
In a long-term collaboration with the Han group in the Computer Science department at Clark, we develop computational methods to extract thermodynamics and kinetics information from molecular dynamics simulations. The methods developed in my group include the MaxFlux reaction-path algorithm combined with the probabilistic roadmap motion planning method, graph representation of protein free-energy landscape, and diffusion maps with a hybrid geometry energy-based kernel.
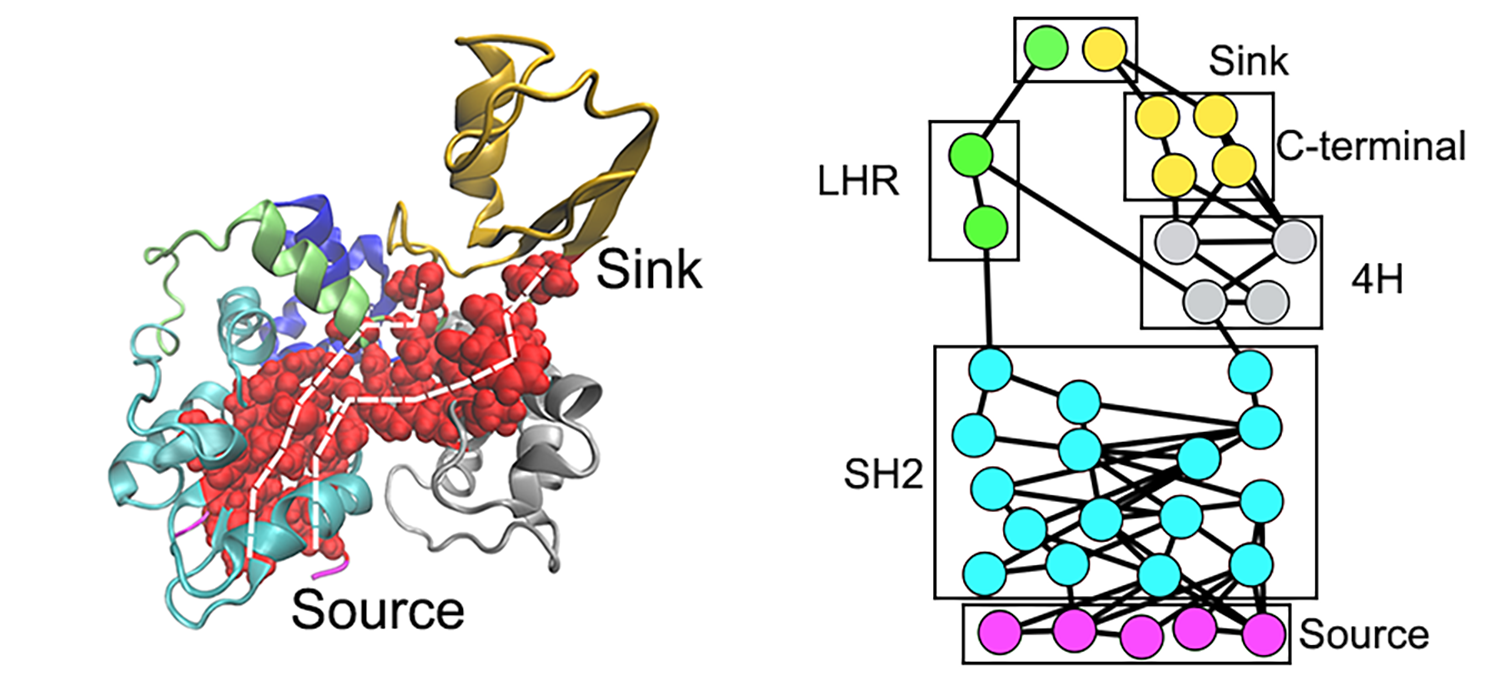 Left panel: Two routes of the allosteric signal transduction; Right panel: Network visualization of allostric signaling pathways
Left panel: Two routes of the allosteric signal transduction; Right panel: Network visualization of allostric signaling pathways
Recently, we developed a subgraph approach for network visualization of optimal and suboptimal allosteric signal transduction pathways. This approach provides a general tool to identify allosteric hotspots and would provide valuable insights into emerging strategies for drug discovery, such as targeted protein degradation.
Computing resources
- Clark University high-performance computing cluster: 320 CPU cores and 5 A30 GPUs.
- Group Exxact GPU workstations.
Funding
NIH grant (No. 1R01-GM088326)