
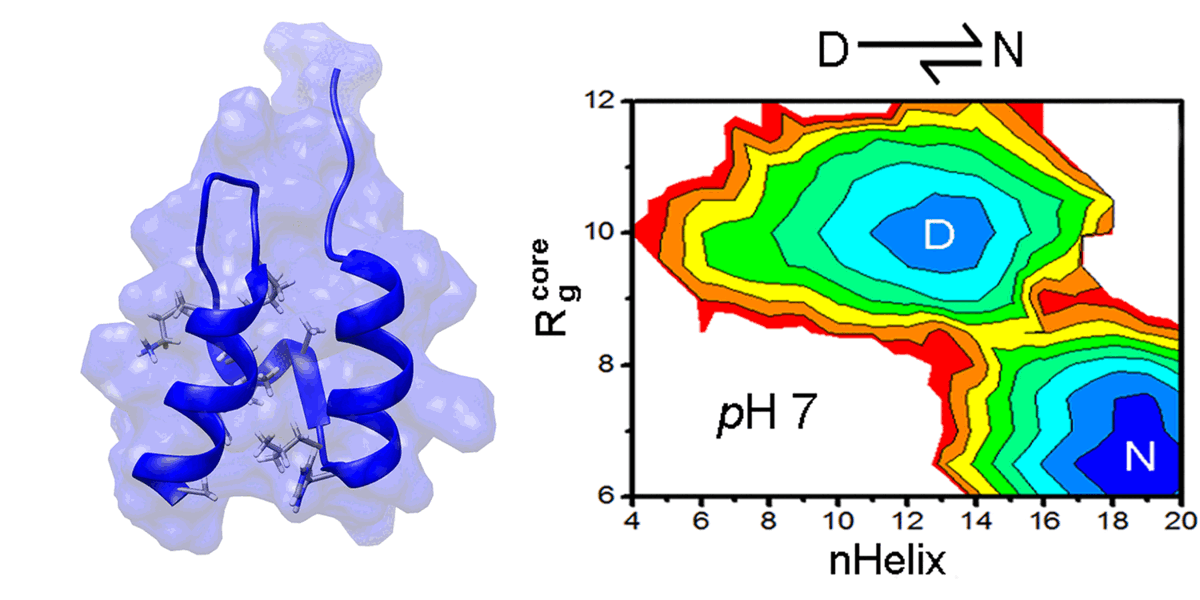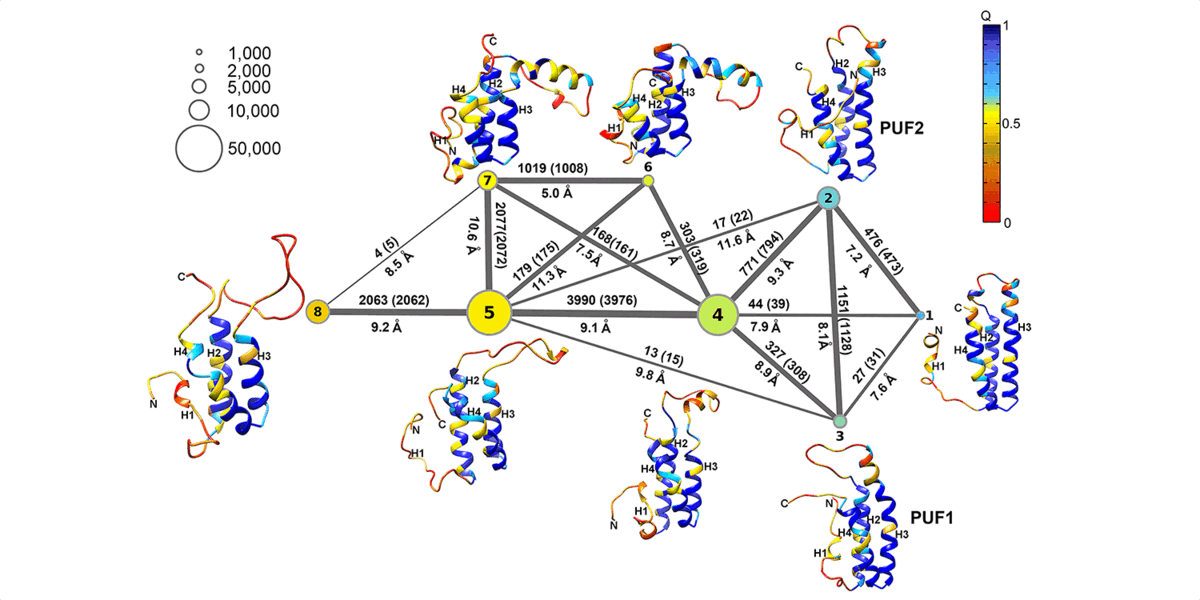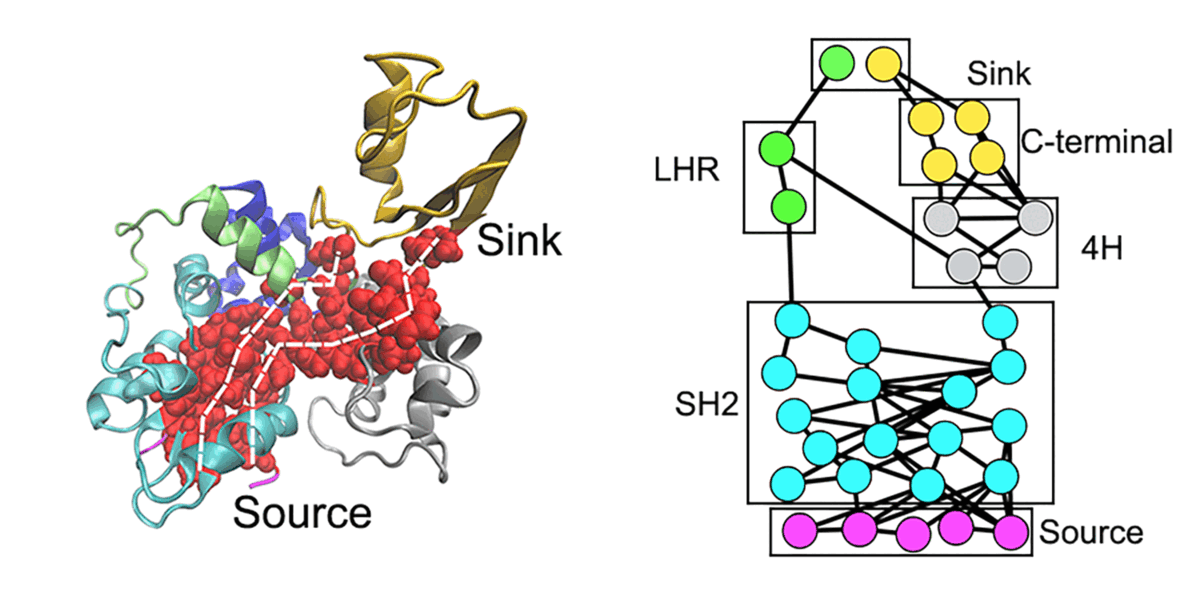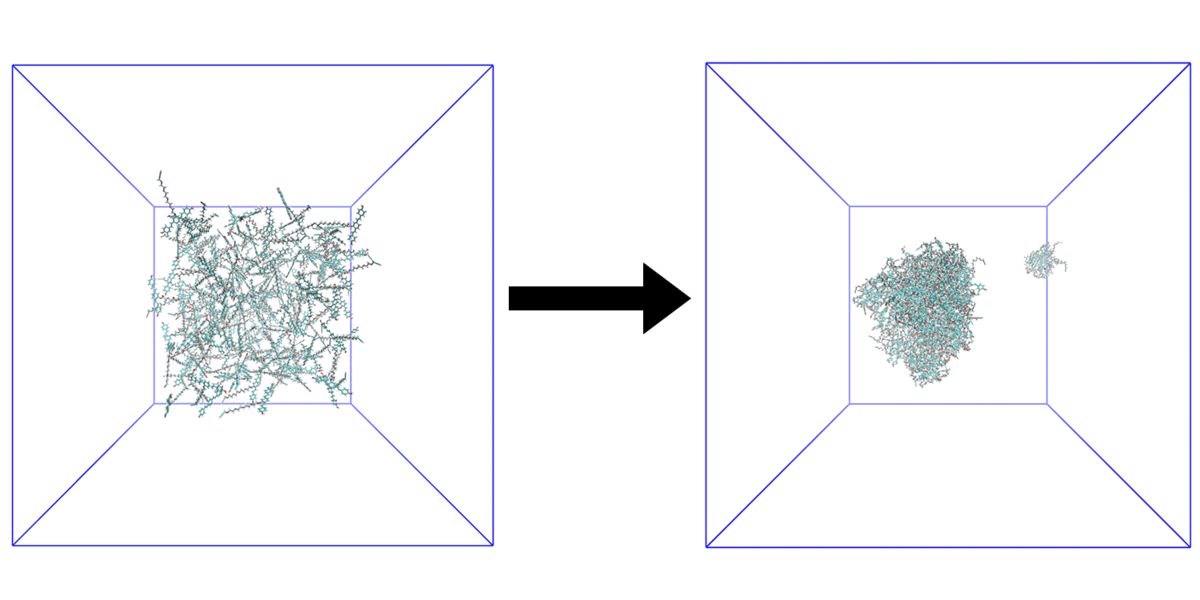
We are interested in computer simulations of protein conformational change, protein-protein interaction, protein-membrane interaction, and protein-small molecule interaction as well as structure-based ligand design. In recent years, our research focuses on the protein allosteric effect, intrinsically disordered proteins, and amyloid aggregate-membrane interactions. We also develop new methodologies for locating protein folding pathways and allosteric signal transduction hotspots using graph theoretical approaches.
We are actively recruiting graduate students. Those with a strong background in physical chemistry or biochemistry and interested in solving biophysical problems using computational approaches are highly encouraged to apply.